sgRNA设计方法及靶基因细胞敲除的应用
2024-09-18
来源: drugdu
 2,528
2,528
基于CRISPR经典的基因敲除系统包含sgRNA序列和Cas蛋白,sgRNA序列长度一般为20个核苷酸。常用于DNA双链切割活性的Cas蛋白包括Cas9和Cas12。由于不同Cas蛋白识别的PAM序列,切割效率及特异性不同,本文将以Cas9蛋白为例详细介绍sgRNA的设计,质粒构建及细胞敲除过程。1.sgRNA设计1.1 基于基因名称的sgRNA设计可通过将基因的名称提交给在线软件完成,目前常用的仅通过输入基因名称就可完成sgRNA设计的在线工具有CHOPCHOP(https://chopchop.cbu.uib.no/)[1], CRISPick(https://portals.broadinstitute.org/gppx/crispick/public)[2-4], 和TKOv3(https://crispr.ccbr.utoronto.ca/crisprdb/public/library/TKOv3/)[5,6]。下面我将以CHOPCHOP软件为例简要介绍其sgRNA设计的具体流程。(1)首先登录CHOPCHOP软件,在Target菜单输入基因名称如TBK1,之后选择合适的物种如‘Homo sapiens’,所用Cas蛋白选择‘CRISPR/Cas9’,sgRNA设计目的选择‘Knockout’,最后选择‘FindTarget Sites’(Figure1)。在选择完基因名称后,需要根据细胞或组织类型选择合适的转录本。

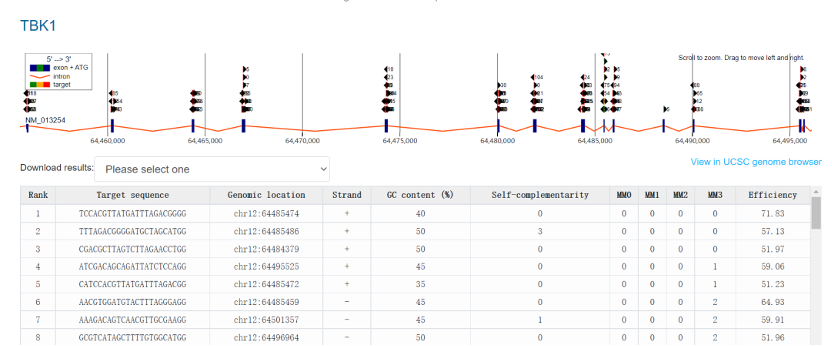
CHOPCHOP运行结束后,将根据sgRNA的切割DNA效率及潜在脱靶情况进行排序(MM0 = 0 mismatches, MM1 = 1 mismatch, MM2 = 2mismatches, MM3 = 3mismatches),我们一般选择切割效率较高且脱靶可能性较低的sgRNA序列,另外由于Cas9在sgRNA引导下可切割双链DNA,并通过造成基因移码突变起到敲除基因的作用;因此,sgRNA序列尽量靶向靠近基因起始密码子ATG下游,最好位于第一或第二外显子上(Figure2)。一般根据上述设计标准,选择2~3条sgRNA进行后续验证。
基于基因序列的sgRNA设计通过输入目标基因序列进行sgRNA序列设计具有更多的灵活性,例如可针对内含子和启动子等基因非编码区域设计,满足上述要求常用的设计软件包括Cas-Designer(http://www.rgenome.net/cas-designer/)和CRISPOR(http://crispor.tefor.net/)[7,8],下面我将以Cas-Designer为例简要介绍该软件的应用方法。(1)进入Cas-Designer界面后,首先选择Cas类型,软件将根据此确定Cas蛋白识别的PAM序列以选择合适的sgRNA,此处我们可选择SpCas9,其PAM序列为NGG。
 将目标序列如FANCM-EXON2以FASTA格式输入,并选择sgRNA长度,由于不同Cas蛋白的所需sgRNA序列长度不同,我们在此可选其默认长度。例如SpCas9蛋白的sgRNA序列长度为20个核苷酸。另外,我们可根据需要选择合适的物种如HomoSapiens后,点击‘Submit’提交。
将目标序列如FANCM-EXON2以FASTA格式输入,并选择sgRNA长度,由于不同Cas蛋白的所需sgRNA序列长度不同,我们在此可选其默认长度。例如SpCas9蛋白的sgRNA序列长度为20个核苷酸。另外,我们可根据需要选择合适的物种如HomoSapiens后,点击‘Submit’提交。
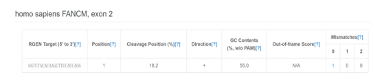
 主要根据“Out-of-frameScore”和“Mismatches”选择合适sgRNA序列,一般sgRNA序列的Out-of-frameScore需大于66,其在基因组中有2个Mismatches的数目应小于2。除此之外,sgRNA中不要出现4个或以上联系“T”核苷酸,以防sgRNA转录终止。基于此我们可通过输入FANCM-EXON2序列:ggtctacacaagcttccaccaggaaggaaatatggtgcagtaagagagtgctttttcttacacctcaggtcatggtaaatgacctttctagaggagcttgtcccgctgctgaaataaagtgtttagttattgatgaagctcataaagctctcggaaactatgcttattgccag。选择候选sgRNA序列如AGGTCATTTACCATGACCTG,ATGGTAAATGACCTTTCTAG和CGGGACAAGCTCCTCTAGAA。值得注意的是当sgRNA第一个碱基为A、C或T,可在sgRNA第一个碱基前面加g以提高sgRNA的转录效率。
主要根据“Out-of-frameScore”和“Mismatches”选择合适sgRNA序列,一般sgRNA序列的Out-of-frameScore需大于66,其在基因组中有2个Mismatches的数目应小于2。除此之外,sgRNA中不要出现4个或以上联系“T”核苷酸,以防sgRNA转录终止。基于此我们可通过输入FANCM-EXON2序列:ggtctacacaagcttccaccaggaaggaaatatggtgcagtaagagagtgctttttcttacacctcaggtcatggtaaatgacctttctagaggagcttgtcccgctgctgaaataaagtgtttagttattgatgaagctcataaagctctcggaaactatgcttattgccag。选择候选sgRNA序列如AGGTCATTTACCATGACCTG,ATGGTAAATGACCTTTCTAG和CGGGACAAGCTCCTCTAGAA。值得注意的是当sgRNA第一个碱基为A、C或T,可在sgRNA第一个碱基前面加g以提高sgRNA的转录效率。
SpCas9所需sgRNA为不包括“NGG”的20个碱基序列。由于SpCas9具有潜在脱靶效应,研究人员目前通过SpCas9蛋白氨基酸点突变的方式构建了SpCas9的各种突变体以提高SpCas9的特异性,其不同突变体特异性高低为evoCas9>> HypaCas9 ≥ SpCas9-HF1 ≈ eSpCas9(1.1) >xCas9 > Sniper-Cas9 > SpCas9,而其相应的DNA切割活性高低为SpCas9 ≥ Sniper-Cas9 > eSpCas9(1.1)> SpCas9-HF1 > HypaCas9 ≈ xCas9 >>evoCas9[9]。为更好的选择合适的sgRNA和SpCas9突变体组合,我们可利用DeepSpCas9variants,验证上述选择sgRNA序列在何种SpCas9突变体中可同时确保较高的切割特异性和有效性[9]。例如,当我们输入FANCM-EXON2序列后发现靶向FANCM的sgRNA:AGGTCATTTACCATGACCTG,ATGGTAAATGACCTTTCTAG和CGGGACAAGCTCCTCTAGAA,依然在Sniper-Cas9有较高的切割活性。由于Sniper-Cas9比SpCas9特异性高,为此我们可以选择Sniper-Cas9与上述sgRNA联用进行基因敲除。推荐阅读文章链接:国内基因编辑疗法突破不断,即将步入高速发展期
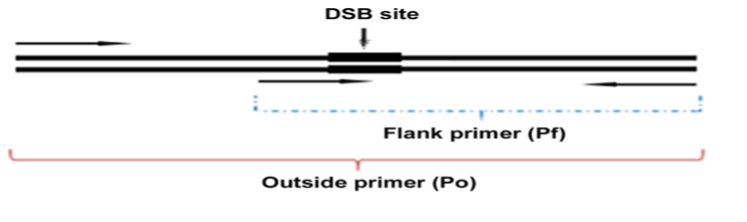
2.sgRNA载体构建sgRNA可构建到LentiCRISPRv2 或lentiGuide-Puro载体中,LentiCRISPRv2慢病毒载体包含Cas9蛋白因此可直接用于敲除实验,lentiGuide-Puro慢病毒载体不含Cas9蛋白,因此该载体需转入到已表达Cas9蛋白的细胞中,其携带的sgRNA方可发挥作用。sgRNA构建到上述载体的详细步骤可参照张峰课题组的protocol。载体构建完成可用U6promoter 通用引物进行测序,验证sgRNA是否成功插入载体。3.病毒包装和细胞感染LentiCRISPRv2 或lentiGuide-Puro慢病毒包装方法可参照addgene提供的protocol进行。慢病毒生产所需包装细胞为293T,所需病毒包装辅助载体为psPAX2和pMD2.G。将收集的病毒利用addgene提供的protocol对目的细胞进行感染并用抗生素(如puromycin)对阳性细胞进行筛选,筛选出的阳性细胞一般需要继续培养7天以确保sgRNA可将目的基因敲除。4.基因敲除鉴定及单细胞克隆筛选基因敲除效率可以用westernblot进行鉴定,或通过提取细胞基因组DNA利用3引物PCR的方法进行验证(Figure6)[10]。由于sgRNA介导的基因敲除主要通过移码突变,因此敲除效率验证一般不用基于mRNA的PCR方法。若在转染sgRNA和Cas9的混合细胞中,经验证发现其基因敲除效率较高。为消除细胞的异质性,可对混合细胞进行单克隆筛选。单细胞克隆筛选可采用极限稀释或流式分选的方法,可参照Corning或 Invitrogen公司提供的具体实验方法操作进行或。由于sgRNA可造成基因组永久性片段缺失,因此也可将具有较高切割效率的sgRNA片段连接到具有荧光标记且表达Cas9的载体上,随后将上述载体瞬时表达到目的细胞,进而通过流式细胞分选将高表达GFP的单细胞分选出来,经扩增培养及基因敲除鉴定以获取敲除效率较高的单细胞克隆。由于该方法是通过瞬时表达CRISPR/Cas9元件,因此经长时间细胞培养后,细胞将不在含有Cas9蛋白及sgRNA序列。
 恢复实验由于sgRNA具有潜在的脱靶效应,为确保目的基因的敲除表型并非脱靶效应导致,除设计多条有敲除效果的sgRNA进行验证外;一般还会通过慢病毒感染的方式恢复目的基因的表达,以观察基因敲除所导致的相应表型是否会被逆转。(1)若敲除细胞是通过慢病毒感染CRISPR/Cas的方法获得,其进一步通过慢病毒重新转入的目的基因的序列中,其被sgRNA识别的碱基序列需要进行同义替换,或同义突变sgRNA识别所需的PAM序列;通过上述方法,敲除细胞原有的CRISPR/Cas9元件将不会识别过表达的基因序列,进而使敲除的基因表达恢复。(2)若敲除细胞是通过瞬时表达荧光标记的CRISPR/Cas载体获得,那么慢病毒携带的目的基因片段则无需进行任何同义替换。总结基于sgRNA的基因敲除方法与shRNA或siRNA方法相比序列脱靶性明显降低,尽管sgRNA作用时间稍长(一般需要1周左右)[11];但sgRNA能够造成基因永久性敲除,因此适用于基因敲除细胞的构建。由于通过慢病毒递送CRISPR元件获得的敲除细胞系含有Cas蛋白,因此需要构建相应的对照细胞系,另外为确保敲除效率,针对某一基因一般可设计2~3条不同的sgRNA。除此之外,基因敲除表型最好需要至少2个有敲除效果的sgRNA进行验证。上述基于基因序列的sgRNA设计方法除可用于基因敲除外,还可用于dCas9介导的基因激活、抑制和表观遗传学修饰等所需sgRNA序列。另外,也可通过设计两条sgRNA,利用nCas9(nickaseCas9)实现DNA片段的删除,插入和碱基位点的替换。Cas9具有两个切割活性中心-RuvC和HNH,负责双链DNA的切割。RuvC突变体Cas9(D10A)仅在靶向链上产生切口,而HNH突变体Cas9(H840A)仅在非靶向链上产生切口;nCas9是Cas9(D10A)或Cas9(H840A)。然而利用两条sgRNA和nCas9介导的DNA片段插入和碱基位点突变依赖于同源重组修复(Homologydirectedrepair),而此过程主要发生在分裂细胞的G2期和S期,因此该方法无法对完全分化的细胞如神经细胞,心肌细胞和骨骼肌细胞实现上述基因编辑。但碱基编辑器和先导编辑器的发现可突破上述传统编辑器的限制。
恢复实验由于sgRNA具有潜在的脱靶效应,为确保目的基因的敲除表型并非脱靶效应导致,除设计多条有敲除效果的sgRNA进行验证外;一般还会通过慢病毒感染的方式恢复目的基因的表达,以观察基因敲除所导致的相应表型是否会被逆转。(1)若敲除细胞是通过慢病毒感染CRISPR/Cas的方法获得,其进一步通过慢病毒重新转入的目的基因的序列中,其被sgRNA识别的碱基序列需要进行同义替换,或同义突变sgRNA识别所需的PAM序列;通过上述方法,敲除细胞原有的CRISPR/Cas9元件将不会识别过表达的基因序列,进而使敲除的基因表达恢复。(2)若敲除细胞是通过瞬时表达荧光标记的CRISPR/Cas载体获得,那么慢病毒携带的目的基因片段则无需进行任何同义替换。总结基于sgRNA的基因敲除方法与shRNA或siRNA方法相比序列脱靶性明显降低,尽管sgRNA作用时间稍长(一般需要1周左右)[11];但sgRNA能够造成基因永久性敲除,因此适用于基因敲除细胞的构建。由于通过慢病毒递送CRISPR元件获得的敲除细胞系含有Cas蛋白,因此需要构建相应的对照细胞系,另外为确保敲除效率,针对某一基因一般可设计2~3条不同的sgRNA。除此之外,基因敲除表型最好需要至少2个有敲除效果的sgRNA进行验证。上述基于基因序列的sgRNA设计方法除可用于基因敲除外,还可用于dCas9介导的基因激活、抑制和表观遗传学修饰等所需sgRNA序列。另外,也可通过设计两条sgRNA,利用nCas9(nickaseCas9)实现DNA片段的删除,插入和碱基位点的替换。Cas9具有两个切割活性中心-RuvC和HNH,负责双链DNA的切割。RuvC突变体Cas9(D10A)仅在靶向链上产生切口,而HNH突变体Cas9(H840A)仅在非靶向链上产生切口;nCas9是Cas9(D10A)或Cas9(H840A)。然而利用两条sgRNA和nCas9介导的DNA片段插入和碱基位点突变依赖于同源重组修复(Homologydirectedrepair),而此过程主要发生在分裂细胞的G2期和S期,因此该方法无法对完全分化的细胞如神经细胞,心肌细胞和骨骼肌细胞实现上述基因编辑。但碱基编辑器和先导编辑器的发现可突破上述传统编辑器的限制。
版权著作,未经滴度drugdu.com书面授权,严禁转载,违者将被追究法律责任。
相关News
- 中国细胞生物产业大会与生物医药创新合作大会 CBIC 2026-07-16
- 中国创新药爆单!阿斯利康15亿美元买断舒沃替尼 2026-07-15
- 黄金五年已开启!第二十八届高交会BCE亚洲生物技术与化学工程展览会带你抢占先机 2026-07-10
- 【企业推荐】上海万特万科技有限公司 2026-07-10
- 歌礼制药两款肥胖症新药向FDA递交IND申请 2026-07-09




